We explore global and anharmonic internal motions of large systems on microsecond time scales which are not achievable to standard all-atom molecular dynamics simulations. We use simplified descriptions of biomolecules and represent groups of atoms with single interacting centers. Because we are interested mainly in one-bead models, entire amino-acids and RNA/DNA nucleotides are represented as single beads centered on the positions of the CA and P nuclei. This approach allows us to reduce the number of degrees of freedom and study low-resolution crystal structures as well as large systems such as the ribosome.
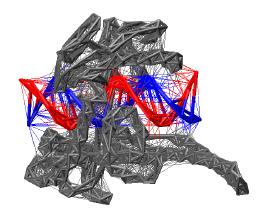
Figure: Schematic representation of the coarse-grained model for the topoisomerase (PDB entry 1A36). Vertices represent CA protein atoms or DNA phosphorus atoms. Thick lines represent the harmonic bonded interactions and thin lines the Morse potentials. The interaction network for protein is shown in grey and for the DNA in red and blue.
These models are an extension of the elastic network models but incorporate anharmonic potentials and distinguish various types of bonded and nonbonded interactions to account for the decay of interactions with distance. The parameterization of internal interactions is crucial for these classes of models and different parameterizations are necessary for different purposes. Our models are parameterized either on the Boltzmann inversion procedure based on the crystals structures of the studied system or on all-atom molecular dynamics simulations in explicit solvent.
We implemented the models into our own software which can be downloaded from Software.