
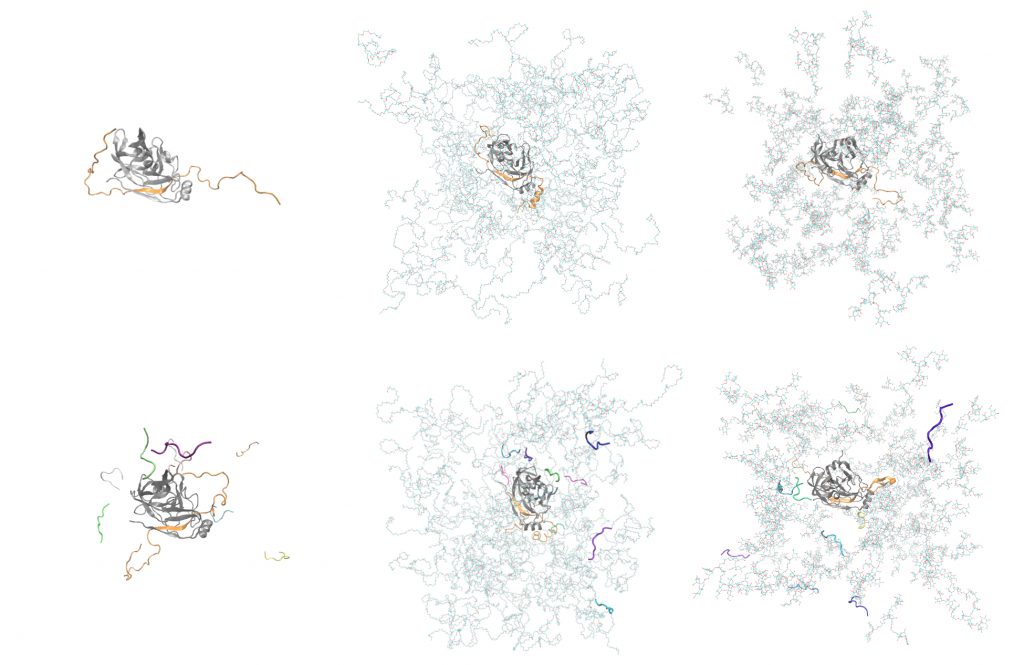
In the OPUS 2017-2021 and Tandems for excellence projects, we conducted molecular dynamics simulations of the NS3/4A protease from Hepatitis C Virus in a crowded environment modelled with three types of crowders: tetramers of Ficoll-like polysucrose and 28-mers of polyethylene glycol (PEG) in atomistic resolution, and the coarse-grained (CG) crowders of our design (all published in [1-4]).

The starting structures of PEG crowders were sampled from a series of molecular dynamics simulations of a single 28-mer in explicit water, conducted by H. Lee at al.[5]. PEG molecules were parametrised using the standard CHARMM36 force field and partial charges used in the single PEG simulations[5], supplemented with the optimized values for the torsion angles[6].
Ficoll-like polysucrose model was built with four sucrose monomers connected with three glycerol linkers to match the number of atoms in our PEG crowder. Basic components of the structure were parameterized using standard CHARMM36 parameters for sucrose and glycerol; force field parameters for the sucrose-glycerol connections were chosen based on the structural similarity to the di- and tri-saccharides described in the CHARMM force field [3].
To avoid excessive aggregation observed in the simulations with PEG in the standard TIP3P water model, the water-solute interactions were upscaled by the factor of 1.09, based on the optimization performed for the charmm force field[7] . In the latter optimization process, such scaling factor eliminated excessive aggregation of test peptides, and did not to destabilize their native structures[7]. In case of our simulations, the modification of water interactions restored realistic diffusion of PEG crowders [1]. For consistency, it was used in all types of the simulated systems that included explicit water molecules.
The CG model was specifically optimised to be used in atomistic simulation systems – its structure and interactions with other molecules and water are aligned with technical requirements of the NAMD software, and parameterized for the CHARMM force field. Our CG crowder is a spherical structure of a radius corresponding to a folded 28-mer of PEG, built with 42 carbon-sized pseudo-atoms evenly distributed on a surface of a sphere and an additional large pseudo-atom in the center. The central atom ensures structural stability of the crowder – without this atom the crowder would collapse inwards, due to the water pressure outside, and a vacuum inside. The surface atoms are connected with pseudo-bonds modelled with strong Lennard-Jones potential – this solution was introduced because of the limitations of the NAMD program, regarding the distance between four bound atoms. To minimize the interactions between the crowders and protein, all atoms of the crowders are uncharged. To overcome high hydrophobicity arising from the lack of partial charges, artificial hydrophilicity is implemented, in a form of the specified strength of crowder-water interactions. These interactions were optimized in a way that the distribution of water molecules around the CG crowder corresponds to the distribution of water around the PEG crowders [2].
To avoid formation of pseudo-bonds between the atoms of separate crowders, it was necessary to assign each crowder with a separate type of the central atom, and a separate type for the surface atoms. For this reason, for each simulation system one needs to generate a crowder parameter file depending on the number of crowders in a system. A program for generating CHARMM parameter files for the crowders, along with other scripts and files needed to use our coarse-grained crowders in a molecular dynamics simulation, is available on request, in a form of a package of files and a detailed instruction.
In total, simulations for a series of seven types of molecular systems was conducted, containing the NS3/4A protease surrounded with explicit water molecules and ions and in addition:
- all-atom PEG crowders,
- all-atom polysucrose crowders,
- spherical coarse-grained crowders,
- peptide substrates,
- PEG crowders and peptide substrates,
- Ficoll crowders and peptide substrates.
In all simulations the TIP3P water model was used, along with Na+ and Cl- ions. Although in the living cell, the substrate of the NS3/4A protease is the polyprotein, in our simulations the substrate was represented analogously as in laboratory assays measuring the activity of the protease, namely as a short peptide effectively cleaved by NS3/4A[1].
The results of our simulations were described in a series of related publications:
- the first article describing the opposite effects of PEG and Ficoll on the activity and inhibition of the NS3/4A protease[1],
- the study comparing the effects of the presence of two types of crowders, all-atom PEG and the CG crowders, on the dynamics and structure of the protease and its protein cofactor[2],

- the concluding study describing the results of a complete set of molecular dynamics simulations, offering hypotheses explaining different effects of PEG and Ficoll on the activity of NS3/4A[3],
In addition to the original research paper series, we published a review briefly describing different approaches to including the crowded environment in molecular dynamics simulations[4].
References:
[1] Crowded environment affects the activity and inhibition of the NS3/4A protease. A. Popielec, N. Ostrowska, M. Wojciechowska, M. Feig and J. Trylska; Biochimie 176:169-180, 2020 [2] Crowding affects structural dynamics and contributes to membrane association of the NS3/4A complex. N. Ostrowska, M. Feig and J. Trylska; Biophysical Journal 120(17):3795-3806, 2021 [3] Varying molecular interactions explain crowder-dependent enzyme function of a viral protease. N. Ostrowska, M. Feig, J. Trylska; BioRxiv [4] Modeling crowded environment in molecular simulations. N. Ostrowska, M. Feig and J. Trylska; Frontiers in Molecular Biosciences 6:86, 2019 [5] Molecular dynamics studies of polyethylene oxide and polyethylene glycol: Hydrodynamic radius and shape anisotropy. H. Lee, R.M. Venable, A.D. MacKerell Jr, and R.W. Pastor; Biophysical Journal, 95(4):1590–1599, 2008. [6] Parameterization of the charmm all-atom force field for ether lipids and model linear ethers. A. N. Leonard, R. W. Pastor, and J. B. Klauda; J Phys Chem B, 122(26):6744–6754, 2018. [7] Slow-down in diffusion in crowded protein solutions correlates with transient cluster formation. G. Nawrocki, P.H. Wang, I. Yu, Y. Sugita, and M. Feig; J. Phys. Chem. B., 121:11072-11084, 2017.