MINT (Motif Identifier for Nucleic acids Trajectory) can analyze structure of your RNA or DNA molecule meaning: 
– find secondary and tertiary contacts (Watson-Crick, WC-WC, and non-Watson-Crick, non-WC-WC) with their conformations
– compute the energy of stacking interactions between nucleotides using a molecular mechanics approach.
– find pi-stacking pairs
– detect and classify all RNA structural motifs (such loops, bulges, junctions and pseudo-knots) with their specifications.
– outline WC-WC triplexes.
How to download MINT?
If you find MINT useful, please acknowledge its use by citing:
Anna Górska, Maciej Jasiński, Joanna Trylska, MINT: software to identify motifs and short-range interactions in trajectories of nucleic acids, Nucl. Acids Res., 2015, DOI: 10.1093/nar/gkv559.
About MINT
MINT is an automatic standalone tool for analyzing three-dimensional structures of RNA and DNA molecules, their full-atom molecular dynamics trajectories or other conformation sets (e.g. X-ray or NMR-derived structures). You can also draw your structure (using VARNA) and colour it according to the computed parameters.
Output files
MINT produces multiple output files. In all of them nucleotides are described in a manner that allows to identify them unambiguously. We use the representation containing a chain name, a residue name and a residue number. For example: the N|GUA:521 represents residuum number 521 from the chain N, with the name GUA. The values posted in the chain name|residue name:residue number code are derived from the input PDB file. The zipped package contains the following files:
_description – the main file containing the complete description of the structure. The file content starts from the list of all of the used parameters, followed by the list of helices, motifs, triplets and pseudo-knots. One can also find the list of both Watson-Crick (WC-WC) and non-Watson-Crick interactions, with the exact parameters of the hydrogen bonds. Additionally there is a simple dot-bracket representation of the secondary structure that can be used for visualization or energy computation by other tools.
_helices.csv – a file that contains a list of the deceted helices.
_ion_pi.csv – a file that contains a list of the recognized ion-pi interactions (i.e., detected if a phosphorus atom in one nucleotide is placed close to the nucleobase of the second nucleotide) with their interaction energy values.
_motifs.csv – a file that contains list of the all RNA structural motifs.
_nucleotides_eval.csv – a file that contains the number of the WC-WC hydrogen bonds, non-WC-WC hydrogen bonds, Coulomb energy, van der Waals energy and their sum per nucleotide. The content gives the energetic description of the state of every nucleotide.
_pairs.csv – a file that contains list of all detected pairs along with its classification according to edge (WC, Hoosteen, Sugar) and configuration (Cis or Trans).
_pseudo.csv – a file that contains list of the all detected pseudo-knot.
_stacking.csv – a file that contains a list of all recognized stacking pairs with their interaction energy values.
_triplets.csv – a file that contains the number of the recognized triplets.
_MINT.xls – a multiple sheets file that combines the above .csv files.
.pdb files – the analyzed structure with the column containing the beta-factors replaced with: the number of WC-WC hydrogen bonds (_2D.pdb), non-WC-WC hydrogen bonds (_3D.pdb), energy of the Coulomb interactions (_Coulomb.pdb), van der Waals interactions (_VDW.pdb) and their sum (_stacking_sum.pdb). These files may be used for 3D visualizations, e.g., in VMD or Chimera.
.png files – pictures with 2D visualizations of computed parameters from Varna. Naming convention is similiar to the .pdb files.
Docs
Hydrogen bonds and base pairs
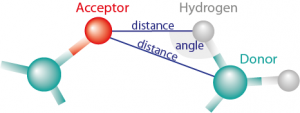
Figure 1: Hydrogen bond is detected when the distance is smaller then cutoff and angle (created by Acceptor, Hydrogen and Donor) is larger than cutoff angle. MINT can measure distance between both Acceptor and Donor, and Acceptor and Hydrogen.

Figure 2: Every nucleotide has three sides/edges that can create hydrogen bonds with another nucleotide.
Figure 3: Yellow spheres correspond to atoms: C1′ and N1 in pyrimidines or N9 in purines. The torsion angle created by these four points determines the configuration of the two nucleotides creating a pair.
0] U2/A116 N3-H3-N1 angle: 150.65 distance: 1.91 | N3-H3-C2 angle: 164.89 distance: 2.48 | O4-H61-N6 angle: 151.98 distance: 1.82 WC/WC/3 Cis
1] C3/G115 N4-H42-O6 angle: 147.3 distance: 2.26 | O2-H22-N2 angle: 168.89 distance: 2.01 | N3-H1-N1 angle: 166.09 distance: 2.08 WC/WC/3 Cis
2] U4/A114 N3-H3-N1 angle: 169.96 distance: 1.83 | N3-H3-C2 angle: 156.01 distance: 2.65 | O4-H61-N6 angle: 151.51 distance: 2.23 WC/WC/3 Cis
3] U5/A113 N3-H3-N1 angle: 167.78 distance: 1.91 | N3-H3-C2 angle: 160.28 distance: 2.5 | O4-H61-N6 angle: 150.16 distance: 2.42 | O2-H2-C2 angle: 140.25 distance: 2.76 WC/WC/4 Cis
0] G0/C118 N2-H22-N3 angle: 168.54 distance: 1.89 WC*Sugar/WC/1 Cis
1] U1/A117 O4-H61-N6 angle: 162.14 distance: 1.71 WC*Hoogsteen/WC*Hoogsteen/1 Cis
2] A12/C80 C2-H2-O2 angle: 167.64 distance: 2.17 WC*Sugar/WC*Sugar/1 Trans
Motifs

Figure 4: A scheme representing several types of structures occurring in RNA with their MINT codes. Pictures generated using Varna applet.
1] G500-C504 -> G541-C545
2] C511-G515 -> C536-G540
3] A520 -> A533
4] G521-C522 -> G527-C528
1] resid 500 to 504 541 to 545
2] resid 511 to 515 536 to 540
3] resid 520 520
4] resid 521 to 522 527 to 528
1] 3-0-16 C8-G110-A109-A108-G107-G106-C90-G89-C25-A24-G23-G22-G21-A20-G19-A18-C17-G16-A15-A14-G13-A12-G11-A10-A9-C8-
2] 4-14 G28-C86-U85-A84-A83-A82-U81-A43-C42-U41-U40-C39-G38-A37-A36-G35-C34-A33-G32-C31-C30-C29-G28-
3] 1-2 C45-G79-U78-G77-C48-A47-A46-C45-
…
Pseudo-knots

Figure 5: An example of a secondary structure of an RNA molecule and its list representation. Red lines correspond to the Watson-Crick interactions creating a pseudo-knot.
G13-C42 A14-U41 A15-U40 G16-C39 C17-G38 G21-C31 G22-C30 G23-C29 U85-A109
Triplets

1] A1014-G987-C1218-
Triplets vmd
1] resid 1014 987 1218
Stacking

n1 n2 Coul VdW sum
G0 U1 (2.62, -6.0, -3.38)
G0 U2 (1.06, -0.3, 0.76)
U1 U2 (3.86, -3.99, -0.13)
base1 base2 OPatom_from_base2 avg_distance coulomb_energy vdw_energy energy_sum
C1336_A1238_O1P [4.37, (-19.49, 44.38, 24.89)] G1077_G1079_O2P [3.81, (10.57, 0.5, 11.07)] G1266_G1268_O2P [3.63, (11.54, 0.43, 11.97)]
Download
MINT project is hosted on GitHub, where it can be downloaded in its newest version: github.com/gvalchca/MINT
finds residues forming helices, pseudo-knots and other motifs, as well as outputs frame numbers and the percentage of time particular conformations appeared in the trajectory.
computes clusters of secondary structure motifs and average motifs along with two-dimensional (2D) and three-dimensional (3D) contacts.
recognizes all Watson-Crick and non-Watson-Crick base pairs and fings frame numbers in which a helix was present, as well as the percentage of the trajectory time the helix occurred.
computes the average number of Watson-Crick and non-Watson Crick hydrogen bonds per nucleotide.
estimates the average stacking energy – van der Waals and electrostatic energies and their sum per nucleotide.
calculates the average secondary structure.
provides inputs for 2D and 3D visualizations of the above computed parameters.
Please cite MINT if you find it useful
Górska A, Jasiński M, Trylska J. MINT: software to identify motifs and short-range interactions in trajectories of nucleic acids. Nucleic Acids Res. 2015 May 29. doi: 10.1093/nar/gkv559 PubMed PMID: 26024667.